Es la más compleja de las enfermedades raras; es, de hecho, un conglomerado de enfermedades que tienen en común el secuestro genético de una de las 11 enzimas requeridas para transformar las cadenas de azúcar en proteínas y posibilitar el metabolismo celular.
Se trata de un grupo de enfermedades metabólicas hereditarias causadas por la ausencia o irregularidades en el funcionamiento de las enzimas encargadas de procesar a los glicosaminoglicanos, azúcares de cadena larga que resultan imprescindibles en la construcción de huesos, cartílagos, tendones, córneas, piel o tejido conectivo. El nombre se debe a que los glicosaminoglicanos eran conocidos hace tiempo bajo la denominación de mucopolisacáridos.
A falta de alguna de las 11 enzimas encargadas de regular estas sustancias, los glicosaminoglicanos se acumulan en las células de sangre y tejidos, causando lesiones que ponen en jaque el desarrollo del organismo y que pueden alterar, en muchos casos, la función cognitiva. Se trata de una mutación genética que afecta solamente a los individuos que heredan el gen defectuoso por parte de ambos padres (la excepción a esta norma la constituye la mucopolisacaridosis tipo II, o síndrome de Hunter, en la que sólo la madre transmite el gen defectuoso al hijo). Cosas de la genética, es posible que padres y hermanos de un niño enfermo no presenten muestras de enfermedad. Los no afectados, sin embargo, pueden transmitir la enfermedad a sus hijos.
Clasificación harto compleja
Las mucopolisacaridosis (MPS) se consideran todas ellas enfermedades de almacenamiento lisosómico; comparten muchos signos clínicos, pero presentan grados muy diversos de gravedad. Puede que sus características no sean evidentes al nacer, pero progresan a medida que el almacenamiento de glicosaminoglicanos afecta a huesos, tejidos y demás órganos. Las complicaciones neurológicas pueden incluir daños a las neuronas así como dolores y deterioro en la función motora. Se han identificado siete tipos clínicos y diversos subtipos de mucopolisacaridosis.
Dependiendo de la gravedad de los síntomas, por ejemplo, la MPS I se divide en tres subtipos causados por un déficit de la enzima alfa-L-iduronidasa. La MPS I H, o síndrome de Hurler, es el más grave. Los enfermos presentan retrasos en el desarrollo antes ya del primer año de vida, y el desarrollo se detiene por completo entre los 2 y los 4 años. El lenguaje puede sufrir limitaciones debido a la pérdida de la audición y al agrandamiento de la lengua. Con el paso del tiempo, las membranas transparentes de la córnea se nublan y la retina se deteriora; el síndrome de túnel carpiano hace pronto su aparición y la función motora se ve muy comprometida. Hígado, bazo y corazón experimentan un crecimiento desproporcionado, los enfermos presentan una respiración ruidosa y padecen infecciones recurrentes de las vías respiratorias o los oídos. Los niños con síndrome de Hurler frecuentemente mueren antes de cumplir los 10 años, por complicaciones respiratorias o cardíacas. La MPS I S, o síndrome de Scheie, es la forma más leve de MPS I. Generalmente los síntomas comienzan después de los 5 años y el diagnóstico se hace a partir de los 10. Estos niños presentan un nivel de inteligencia normal o sólo leves problemas de aprendizaje. Las personas con síndrome de Scheie pueden vivir hasta la edad adulta. En tercer lugar, la MPS I H-S, o síndrome de Hurler-Scheie, tiene un debut entre los 3 y los 8 años. Los niños pueden presentar retraso mental moderado y dificultades de aprendizaje. Las irregularidades esqueléticas y sistémicas incluyen baja estatura, rigidez generalizada y progresiva, médula espinal comprimida, córneas nubladas, pérdida de la audición, enfermedades cardiacas, características faciales toscas y hernia umbilical. Algunos enfermos requieren ventilación mecánica asistida. Su esperanza de vida se sitúa entre el fin de la adolescencia y el inicio de la vida adulta.
Los signos clínicos no suelen ser evidentes al nacer, pero progresan a medida que la acumulación de glicosoaminoglicanos afecta a los diferentes órganos
La MPS II, o síndrome de Hunter tiene dos subtipos clínicos. Los niños que padecen MPS II A, la forma más grave, comparten muchas de las características clínicas asociadas al síndrome de Hurler (MPS I H) pero con síntomas más leves. La muerte a causa de dificultades respiratorias o por insuficiencia cardiaca ocurre por lo general hacia los 15 años. Las características físicas de la MPS II B son menos obvias y progresan a un ritmo mucho más lento. El diagnóstico no se realiza hasta la segunda década de vida. La inteligencia no falla y los problemas esqueléticos pueden ser menos graves, pero el síndrome del túnel carpiano y la rigidez de las articulaciones restringen gravemente el movimiento y la altura de estos niños es menor de la normal. Otros síntomas incluyen la pérdida de la audición, visión periférica pobre, diarrea y apnea del sueño. Aunque las complicaciones respiratorias y cardiacas pueden contribuir a la muerte prematura, los enfermos de MPS II B pueden vivir hasta más allá de los 50 años.
La MPS III, o síndrome de Sanfilippo, está marcado por síntomas neurológicos graves (demencia progresiva, comportamiento agresivo, hiperactividad, convulsiones, sordera, pérdida de la visión y una incapacidad para dormir por espacio de varias horas seguidas). Este trastorno tiende a presentarse en tres etapas. Durante la primera, el desarrollo inicial de las capacidades mentales y motoras causa que los niños muestren una cierta incapacidad en el aprendizaje (2-6 años). Algunos niños nunca aprenden a hablar. El crecimiento se detiene a la edad de 10 años. El Sanfilippo A es el más grave de los trastornos de MPS III y es causado por la ausencia o alteración de la enzima heparínn-N-sulfatasa. Los niños que padecen este síndrome tienen la menor tasa de supervivencia de entre todos los que padecen MPS III.
La MPS IV, o síndrome de Morquio, se estima que puede ocurrir en uno de cada 200 mil nacimientos. Sus dos subtipos resultan de deficiencias de las enzimas N-acetilogalactosamina-6-sulfatasa (tipo A) o beta-galactosidasa (tipo B). Las complicaciones neurológicas incluyen la compresión de los nervios espinales y las raíces nerviosas, cambios esqueléticos progresivos extremos, particularmente en las costillas y el pecho. Puede que los niños que padecen la forma más grave del síndrome de Morquio no sobrevivan más allá de los 30 años.
Los niños que padecen MPS VI, o síndrome de Maroteaux-Lamy, tienen un desarrollo intelectual normal, pero generalmente comparten muchos de los síntomas físicos encontrados en el síndrome de Hurler. Este síndrome es causado por un déficit de la enzima N-acetilgalactosamina-4-sulfatasa y tiene un espectro variable de síntomas graves. Las complicaciones neurológicas incluyen córneas nubladas, sordera, espesamiento de la duramadre (la membrana que rodea y protege el cerebro y la médula espinal).
La MPS VII, o síndrome de Sly, es una de las formas menos comunes de mucopolisacaridosis, estimándose que puede ocurrir en menos de uno por cada 250 mil nacimientos. El trastorno es causado por una deficiencia de la enzima beta-glucuronidasa. Los síntomas neurológicos pueden incluir retraso mental leve o moderado a la edad de 3 años, hidrocefalia comunicante, compresión de los nervios, córneas nubladas y una cierta pérdida de la visión periférica y nocturna.
Sólo se ha descrito un solo caso de MPS IX en la bibliografía científica. El trastorno, causado por un déficit de hialuronidasa, cursa con una formación de masas nodulares de tejido suave.
Tratamiento y grupos de ayuda
Las MPS no tienen cura. El tratamiento médico se orienta a las condiciones sistémicas y a mejorar en lo posible la calidad de vida del enfermo. La terapia física y el ejercicio diario pueden retrasar problemas comunes y mejorar la capacidad de movimiento. Los cambios en la dieta no previenen la progresión de la enfermedad, pero se ha descrito que el consumo limitado de leche, azúcar y productos lácteos ha ayudado a algunos individuos a mantener a raya una mucosidad excesiva. La cirugía para extraer las amígdalas y las adenoides puede mejorar la respiración de los individuos afectados por trastornos de las vías respiratorias y apnea del sueño. Los trasplantes de córnea pueden mejorar asimismo. Además de las terapias de sustitución enzimática y el trasplante medular, científicos auspiciados por el Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares (NINDS) de EEUU han demostrado en modelos animales que la terapia genética puede parar la acumulación de materiales en células del cerebro y mejorar el aprendizaje y la memoria. Los expertos tratan también de identificar los genes asociados a las mucopolisacaridosis. En España, los enfermos y sus familiares disponen de una Asociación de las Mucopolisacaridosis y Síndromes Relacionados, la Asociación para las Deficiencias que afectan al Crecimiento y al Desarrollo y la Federación Española de Asociaciones de Enfermedades Raras.
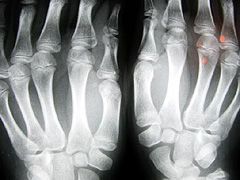
Un equipo de investigadores del Hospital Universitario Virgen del Rocío de Sevilla ha identificado una mutación del gen P533R como causa de mucopolisacaridosis familiar tipo I. Germán J. Rodríguez Criado y sus colaboradores aseguran que esta mutación origina una deficiencia en la alfa-L-Iduronidasa, necesaria para la degradación del heparín sulfato y del dermatín sulfato, «de ahí que estos glucosaminoglucanos se depositen en los lisososmas», añade el experto.
Los investigadores estudiaron los casos de tres pacientes mauritanos, hijos de padres sanos consanguíneos en tercer grado, con un cuadro clínico muy similar, caracterizado por un comienzo de los síntomas al año de vida. Posteriormente desarrollaron facies toscas, opacidad corneal, tumefacción articular, hernia inguinal gigante, marcado retraso pondoestatural, discreto retraso mental, hepatomegalia y osificación defectuosa (disóstosis) con anomalías de la estática y de la marcha.
Los tres casos debieran incluirse, desde el punto de vista fenotípico, entre las MPS tipo I H/S, pues si bien presentaban algunos signos del tipo IH como la talla corta y la disóstosis múltiple, no mostraron un retraso mental importante como ocurre en la enfermedad de Hurler. Los hallazgos exploratorios y de las pruebas complementarias fueron similares a los descritos ampliamente en la bibliografía médica, «salvo la voluminosa hernia umbilical que presentaban los tres hermanos», explica Rodríguez. Las anomalías cerebrales halladas mediante resonancia magnética cerebral no fueron específicas de la MPS IH/S. La sordera fue de patogenia mixta, común en los enfermos con MPS IH/S.




